
Tumour suppressor genes act to inhibit cell proliferation and growth, with the goal of preventing tumours from developing. The major tumour suppressors are RB, TP53, APC, E-cadherin, CDKN2A, TGF-β, PTEN, VHL and STK11.
Two-Hit Hypothesis and Haploinsufficiency
Before learning about tumour suppressors, there is an important concept regarding neoplasia to discuss. Tumour suppressor genes were first described by Knudson, where he hypothesized that in order for a tumour suppressor gene to become dysregulated, they would have to experience “two hits”. In other words, both alleles of a tumour suppressor gene must be mutated (a “hit”) in order for neoplasia to develop. These mutations can occur via point mutations, deletions, mitotic recombination, or other mechanisms. There is also a possibility for DNA methylation of the alleles to play a role, by silencing that allele’s expression.
However, more recent studies have shown that neoplasia and poor tumour suppressor function can develop even with only one “hit”. This has been termed haploinsufficiency. Having one mutated allele can result in reduced amounts of protein to the point where they are unable to maintain homeostasis, or can give rise to a dominant-negative protein. Dominant-negative proteins are proteins that do not have the activity of the original protein, but still bind to the same receptors. Thus, they prevent binding of the active protein, reducing tumour suppressor activity. Through these mechanisms, even one “hit” can be enough to lead to neoplasia.
RB
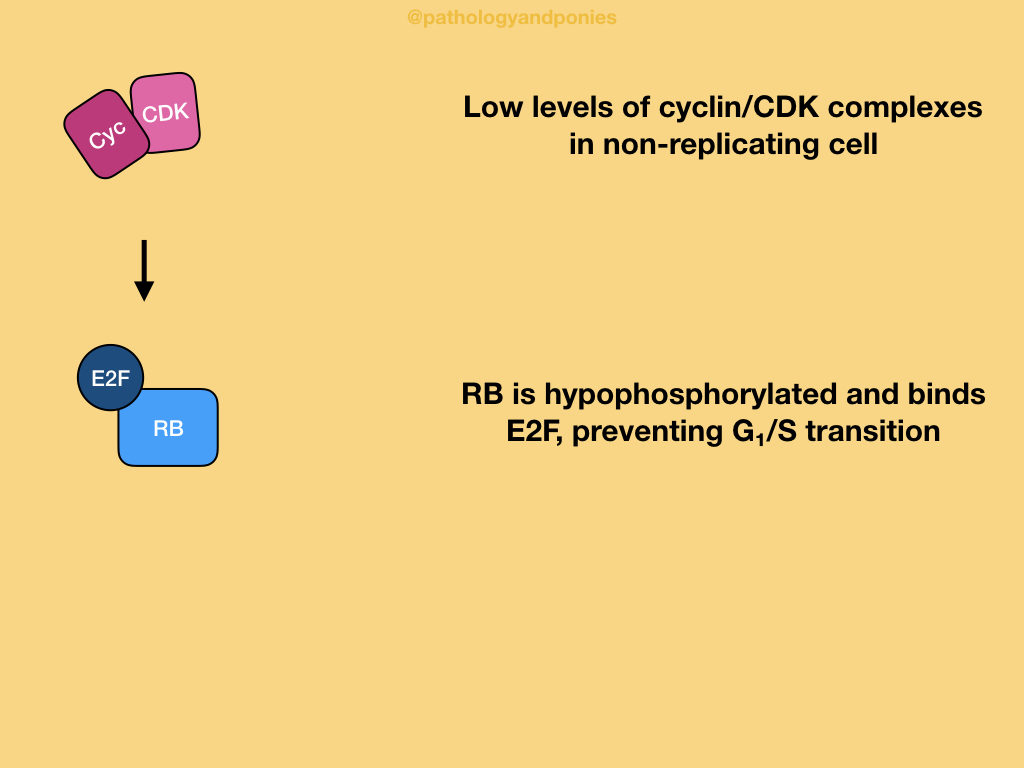
Retinoblastoma protein is considered the governor of proliferation, as it is the most important negative regulator of the G1/S cell cycle transition. The active state of RB is the hypophosphorylated state, which prevents cells from undergoing G1/S.
Homeostasis and Activation
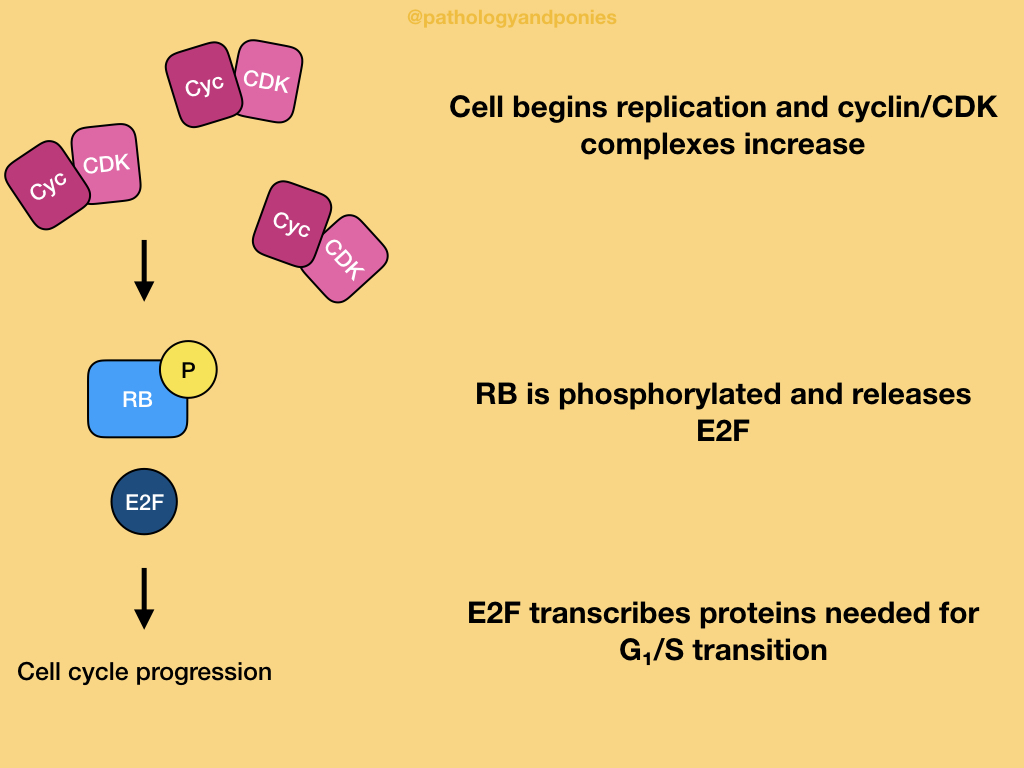
In a normal cell undergoing the cell cycle, high levels of CDK4/cyclin D, CDK6/cyclin D and CDK2/cyclin E lead to hyperphosphorylation of RB, leading to its inactivation. Inactive RB releases E2F transcription factors that drive gene expression for things needed to proceed through G1/S transition. This balance between active and inactive RB is driven by growth factors increasing CDK/cyclin activity and growth inhibitors increased CDK inhibitor activity. Ultimately, RB is the integration point for these signals, giving it an important role in the cell cycle.
Loss of Function
There are three major ways RB’s function can be compromised:
- Loss-of-function mutations in RB alleles, preventing it from working properly.
- Gain-of-function mutations in upregulators of CDK/cyclin activity.
- Loss-of-function mutations in inhibitors of CDK activity.
- Viral oncoproteins that inhibit RB, such as E7 in papillomavirus.
In all of these cases, RB is inactivated and releases E2F, allowing the cell to progress through the cell cycle. Thus, neoplasia develops.
TP53
TP53 is considered the guardian of the genome, due to its ability to regulate the cell cycle, DNA repair, senescence and apoptosis. In fact, it is the most frequently mutated gene in human cancers.
The TP53 gene produces the transcription factor p53, which binds DNA and causes transcription of hundreds of genes. These genes have a variety of functions, including cell cycle arrest, apoptosis and altering metabolism.
Homeostasis and Activation
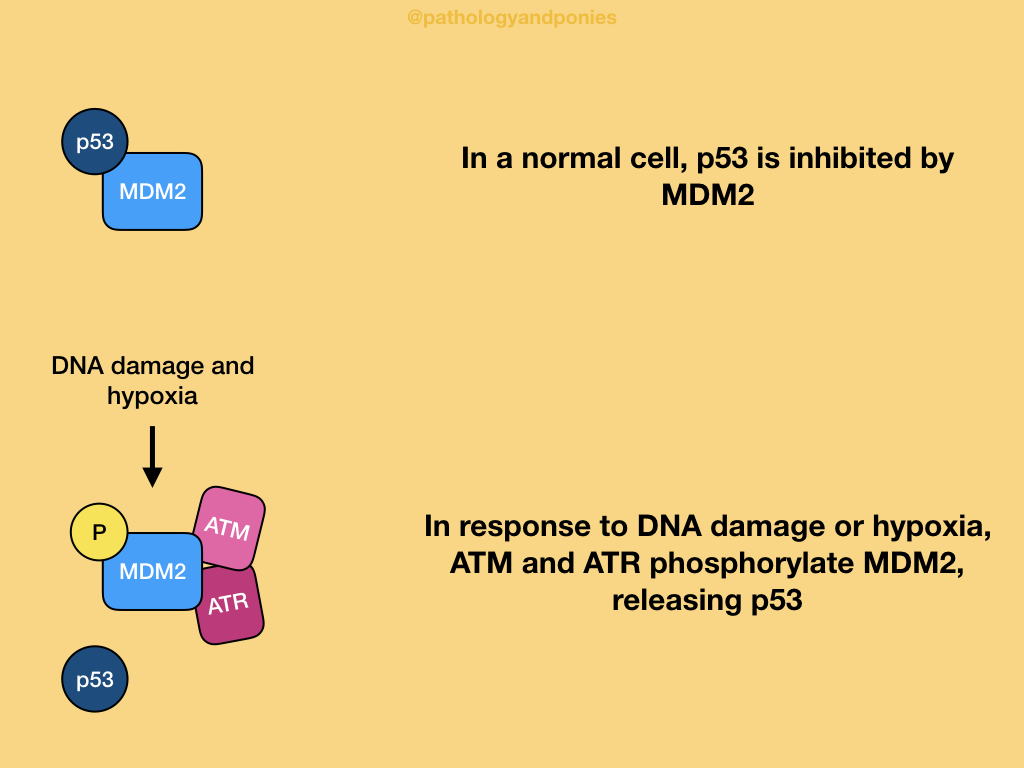
In a normal cell, p53 is inhibited by MDM2, which ubiquitinates p53 to target it for removal. p53 is activated by two main types of cellular stress: DNA damage and hypoxia, and oncogenic stress.
With DNA damage or hypoxia, two protein kinases called ATM and ATR are triggered. These enzymes phosphorylates MDM2, disrupting the binding between the two proteins. This prevents MDM2’s degradative activity, and allows p53 levels to increase within the cell.
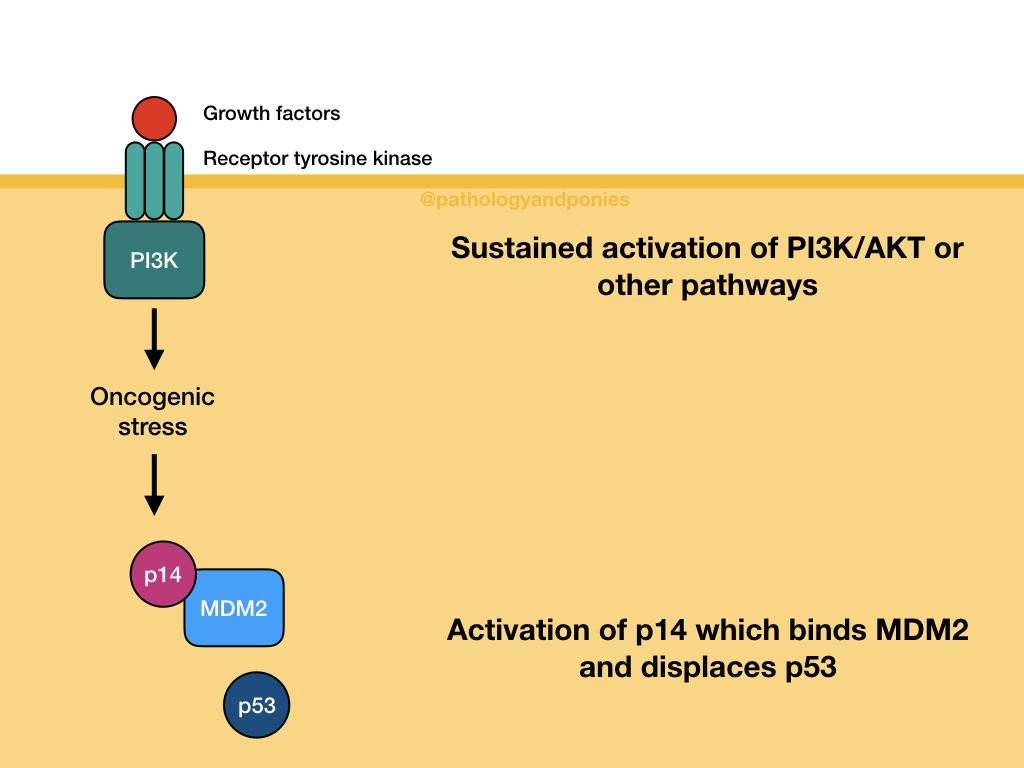
Sustained growth factor signalling through pathways like MAPK or PI3K/AKT leads to cellular stress. This stress leads to activation of p14/ARF, which binds MDM2 and displaces p53. Once again, this allows p53 levels in the cell to increase.
After activation, p53 binds to DNA and transcribes genes relating to cell cycle arrest, apoptosis and altered metabolism. This wide variety of protein production results in several possible outcomes for the cell.
Transient Cell Cycle Arrest
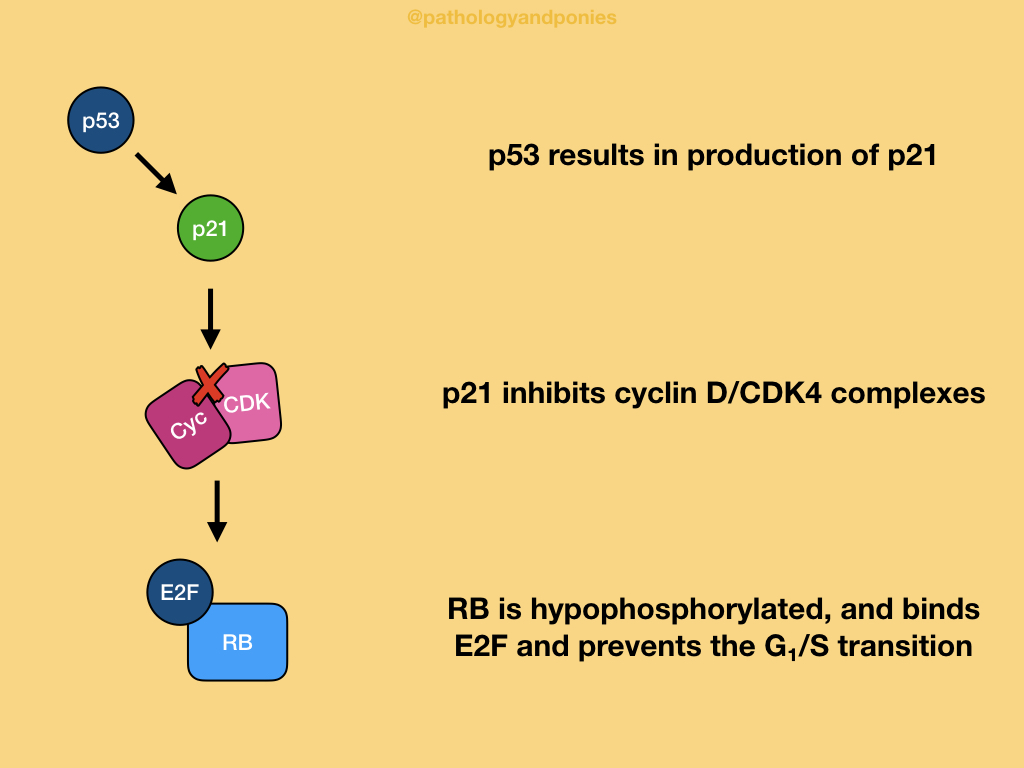
p53-mediated cell cycle arrest occurs late in G1, due to p53’s transcription of CDKN1A leading to production of p21. p21’s main role is to inhibit CDK4/cyclin D, to maintain RB in its active, hypophosphorylated state with no release of E2F. Thus, the progression through the G1/S transition is blocked.
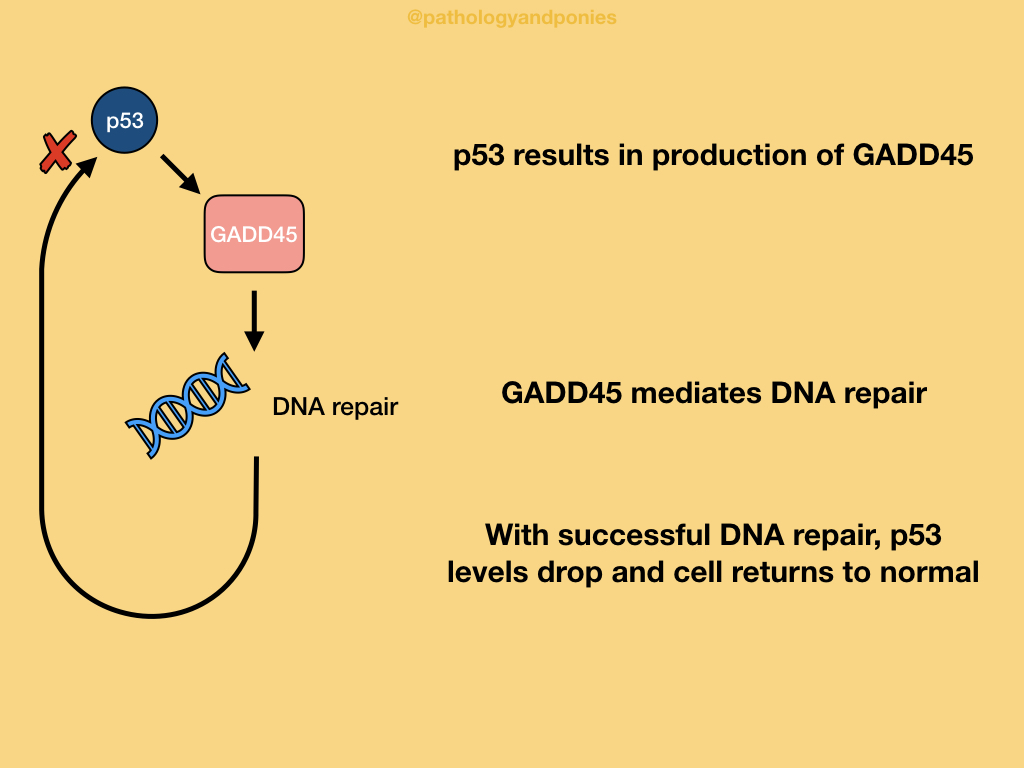
In order to aid in DNA repair during this pause in the cell cycle, p53 induces production of proteins like GADD45 that increase DNA repair. With successful repair, the DNA damage signals that led to p53 activation are decreased, causing reactivation of MDM2 and degradation of p53. Once enough p53 is removed, the cell can return to a normal state.
Senescence
Senescence is a state of permanent cell cycle arrest, which has a somewhat unclear pathogenesis at this time. Currently, it has been theorized that epigenetic changes silence the genes involved in the G1/S transition, preventing the cell from replicating. It is also thought that these epigenetic changes are induced by p53-dependent transcription of genes.
Apoptosis
The most “aggressive” result of p53 activation is apoptosis. p53 increases the transcription of pro-apoptotic BCL proteins like BAX and PUMA, to cause intrinsic apoptosis in the cell.
Determining which outcome occurs for the cell is thought to be related to the level of p53. Lower levels of p53 are thought to induce cell cycle arrest, and if the DNA repair fails, p53 levels will continue to rise. These higher amounts of p53 then result in senescence or apoptosis, depending on whether or not epigenetic processes occur.
Loss of Function
The function of TP53 is most commonly lost due to mutations in the TP53 gene, and may occur with either two “hits” or one “hit”. Loss of function can also occur with mutation of MDM2, leading to overexpression of the protein and subsequently increased degradation of p53. Viral oncogenes can also affect p53, such as E6 protein from papillomavirus binding and degrading p53.
Without p53 function, the cell cycle is not blocked when DNA damage arises. This results in division of cells with mutated, broken, or otherwise disrupted DNA, and the development of neoplasia.
APC
Adenomatous polyposis coli protein is an important tumour suppressor gene linked to colonic neoplasia in humans. It functions by downregulating growth-promoting signalling pathways. Fundamentally, the major role of APC is restricting β-catenin activity.
Homeostasis and Activation
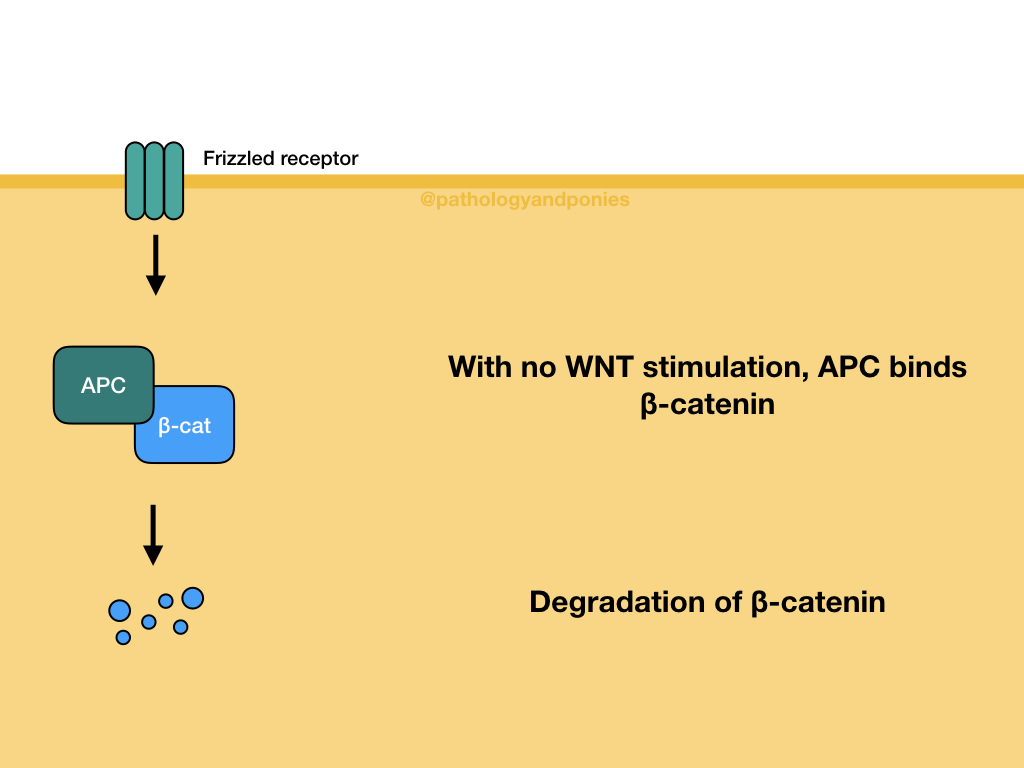
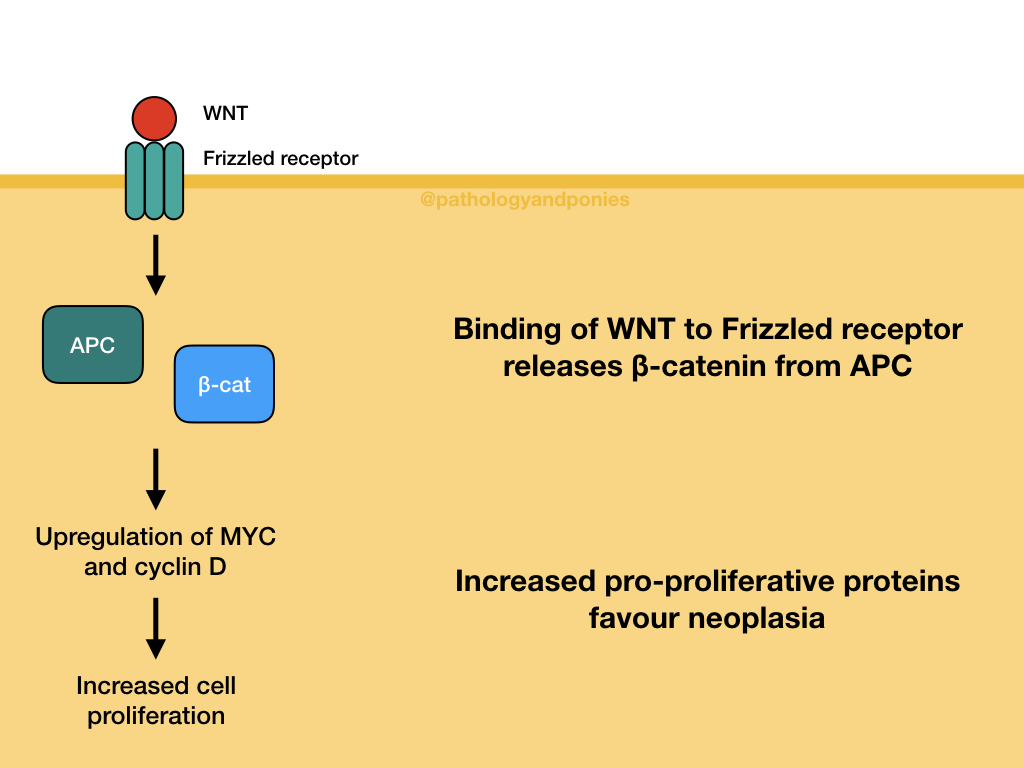
APC is part of the WNT signalling pathway, which controls cell growth and differentiation in embryogenesis. As mentioned, APC’s role in this pathway is regulation of β-catenin activity. In a normal cell without WNT activation, APC forms a destruction complex that degrades β-catenin.
When WNT molecules bind to Frizzled family receptors on the cell surface, the formation of the destruction complex is blocked, stabilizing β-catenin. β-catenin is then able to move into the nucleus and form a transcription complex that upregulates the expression of MYC and cyclin D.
Loss of Function
APC is generally considered to need “two hits” to give rise to neoplasia. This can occur through inherited conditions, like in familial adenomatous polyposis in humans, or through acquired mutations. 70-80% of non-hereditary colorectal carcinomas have APC mutations, so it is very closely linked with colon cancers!
Cells without functional APC behave as if WNT signalling is constantly occurring, leading to very high levels of MYC and cyclin D, promoting cell proliferation. Thus, neoplasia develops.
It is also important to note that this type of signalling can occur without APC mutations, if β-catenin is mutated. Some mutations can prevent β-catenin from binding to APC, thus leading to constant signalling. This type of gain-of-function mutation is notably seen in some hepatocellular carcinomas.
E-Cadherin
E-cadherin is a protein on the cell surface that maintains intercellular connections, particularly in epithelial cells.
Homeostasis and Activation
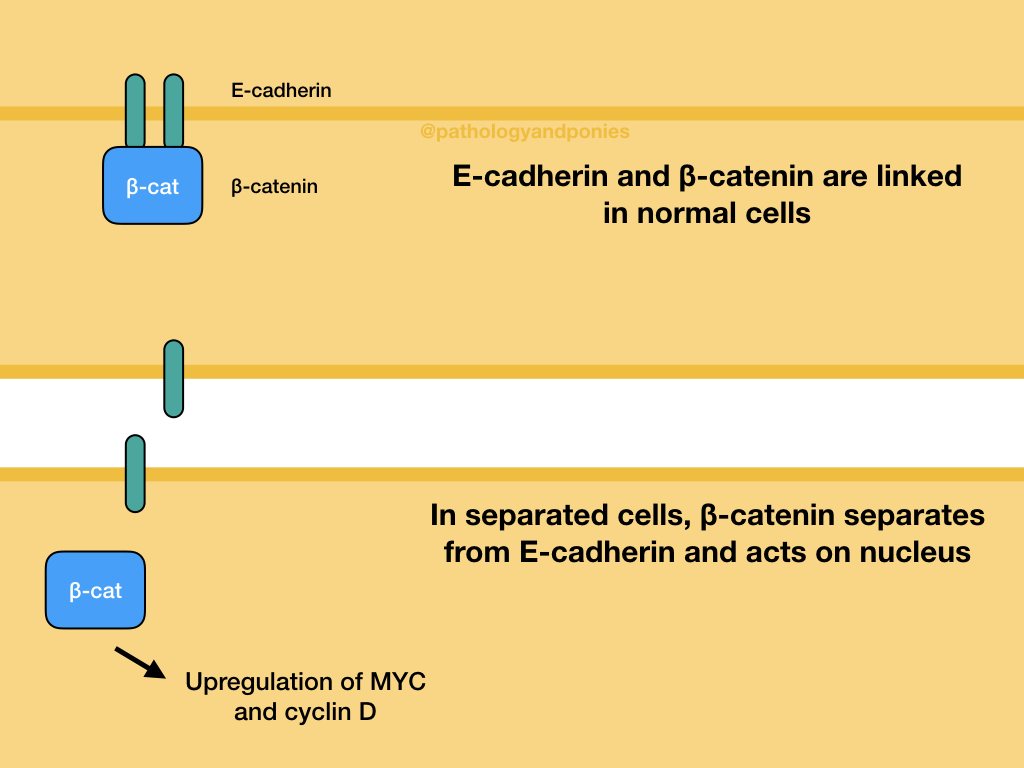
In a normal cell, E-cadherin’s cytoplasmic surface is bound by β-catenin, and tells the cell that it is attached to its neighbour. When there is injury and loss of contact between cells, β-catenin separates from E-cadherin, and acts in the same way as in WNT signalling. When the wound heals, E-cadherin attachments result in β-catenin becoming sequestered at the E-cadherin cytoplasmic surface, re-establishing the connection and preventing further β-catenin signalling. The interaction between E-cadherin and β-catenin is the basis for “contact inhibition” of cells.
Loss of Function
If either E-cadherin or β-catenin are mutated, the cell may proliferate in spite of having adequate intercellular contacts. This is a common finding in carcinomas. E-cadherin mutations can also increase the capacity of the cell to metastasize, because the cells have weakened connections to each other, allowing them to break off and spread to distant sites.
CDKN2A
CDKN2A encodes two of the major regulators of the cell cycle: p16/INK4a and p14/ARF. p16/INK4a blocks phosphorylation of RB protein by CDK4/cyclin D, to prevent G1/S transition. p14/ARF inhibits MDM2, leading to activation of the p53 pathway. Mutations in CDKN2A are relatively common in tumours, due to their control over the cell cycle and proliferation. Specifically, loss-of-function mutations in p16 and gain-of-function mutations in p14 will enhance neoplastic activity.
TGF-β
Transforming growth factor-β‘s main life goal is inhibiting cell proliferation. It does this through stimulating SMAD proteins.
Homeostasis and Activation
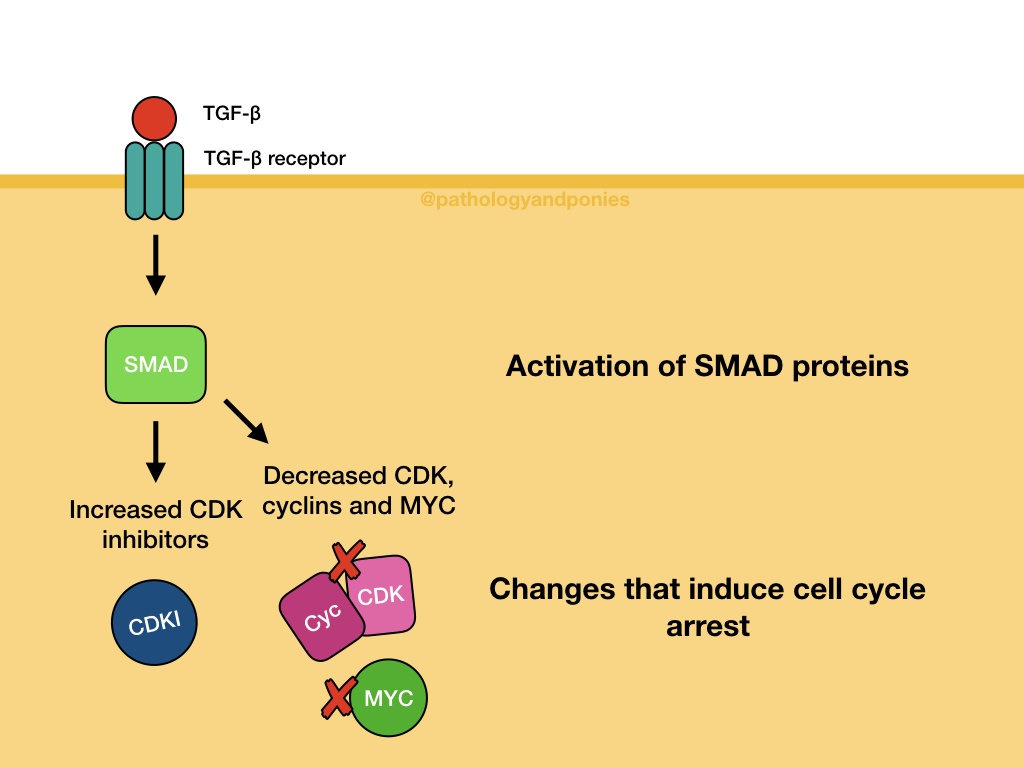
In a normal cell, TGF-β binding to TGF-β receptors activates SMAD proteins, which activate antiproliferative genes and inhibit cell growth genes. For example, SMADs might increase the production of CDK inhibitors, and decrease production of MYC, cyclins and CDKs. Ultimately, these changes decrease RB phosphorylation, and induce cell cycle arrest. The two major regulators of this pathway are SMAD7, which has inhibits phosphorylation and activation of other SMADs, and SMURFs, which can bind to and degrade SMADs and activated receptors.
Loss of Function
Any component of the TGF-β pathway can become mutated, and prevent the normal antiproliferative function of the pathway. In some tumours, TGF-β receptors are mutated, while in others, SMAD proteins are mutated. Mutations can also occur in the products of TGF-β signalling, such as mutations that prevent p21 function or increase MYC function.
PTEN
Phosphatase and tensin homologue (PTEN) is a phosphatase found on cell membranes. Its main function is acting as a brake on PI3K/AKT signalling, thus acting as a tumour suppressor. When PTEN becomes mutated or epigenetically silenced, PI3K/AKT signalling can occur constantly, leading to neoplasia.
VHL
Von Hippel-Lindau protein is part of the ubiquitinating process, which tags proteins for degradation in the proteasome.
Homeostasis and Activation
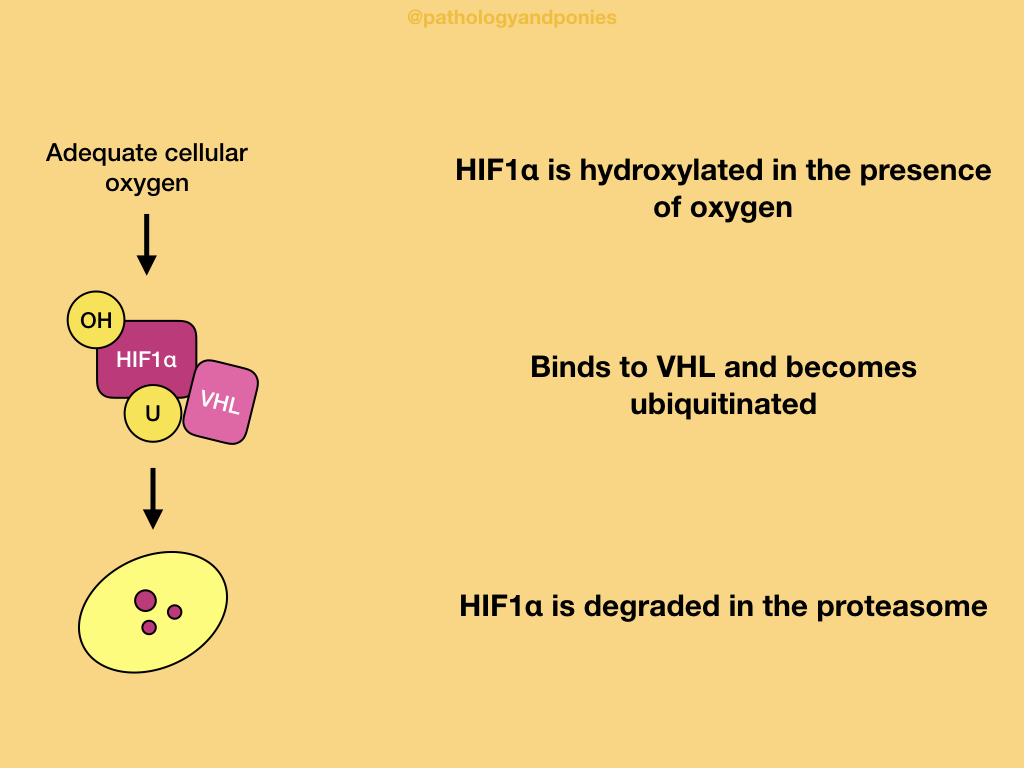
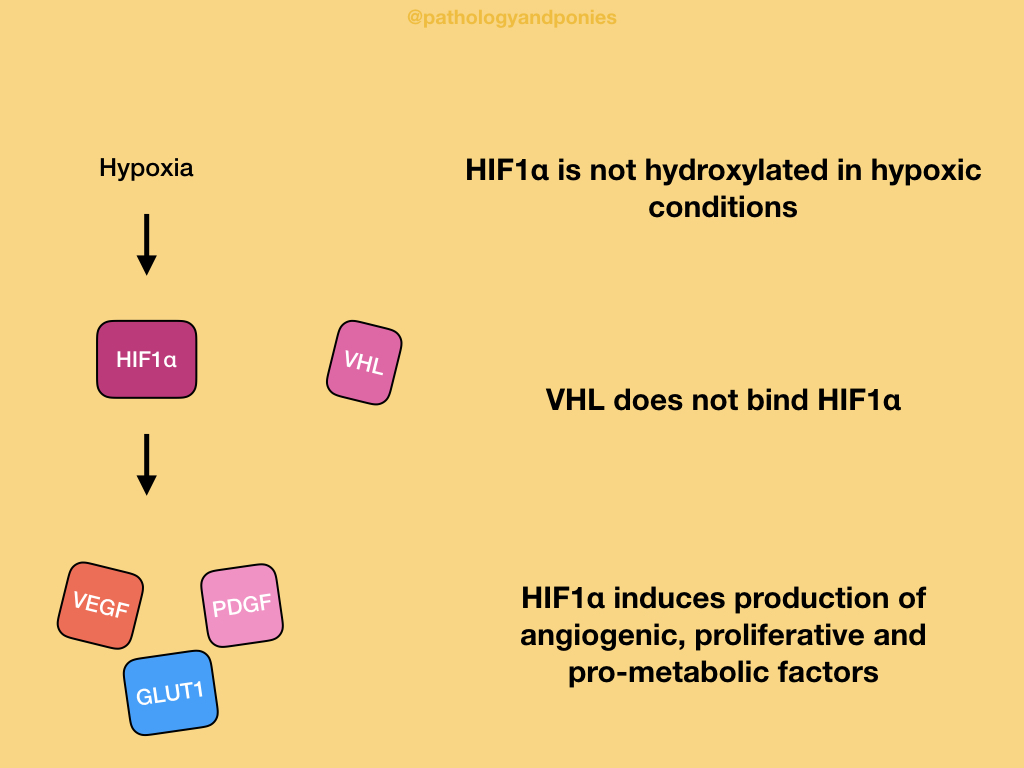
In a normal cell with adequate oxygen, hypoxia-inducible transcription factor 1α (HIF1α) is hydroxylated and binds to VHL. This ultimately causes HIF1α to be ubiquitinated and degraded.
In a hypoxic environment, HIF1α is unable to be hydroxylated, because the process requires oxygen. VHL does not recognize unhydroxylated HIF1α, so the protein accumulates in the cell. HIF1α activates the genes that produce VEGF, PDGF and GLUT1. VEGF induces angiogenesis, PDGF induces cell proliferation, and GLUT1 is a glucose transporter that contributes to the Warburg effect. These factors have a combined effect of increasing the cell’s resistance to hypoxia.
Loss of Function
With loss-of-function mutations in VHL, HIF1α does not get degraded, even with adequate oxygen levels. Thus, the HIF1α-induced genes are continuously activated, leading to increased stimulus for cellular growth and angiogenesis, which favours neoplastic change.
STK11
STK11 is a serine/threonine kinase the regulates cellular metabolism, including glucose uptake, gluconeogenesis, protein synthesis and lipid metabolism. With loss-of-function mutations, these metabolic processes are not adequately regulated, increasing their function and favouring neoplasia.
Zachary JF. Pathologic Basis of Veterinary Disease, Sixth Edition.
Kumar V, Abbas AK, Aster JC. Robbins and Cotran Pathologic Basis of Disease, Tenth Edition.



